





SYSTEM 参数
SYSTEM可以理解为本次计算任务的名称,可随意取。
ISMEAR 参数
不同的值对应着不同的展宽方法:
※ 对于半导体与绝缘体,取值绝对不能大于0,一般用0;
※ 对于K点少的半导体和绝缘体,只能使用0;
※ 对于所有体系,想要获得更为精确到结果时选取-5,且K点数目需要相应地大于等于3;
※ 对于金属,一般取大于等于0的数值;
※ 使用 ISMEAR = 0 即可满足大部分体系的计算。
SIGMA 参数
SIGMA的取值和ISMEAR相关联:
※ 如果ISMEAR=-5,SIGMA的值可以忽略;
※ 对于金属ISMEAR=1/0、非金属ISMEAR=0时,一般SIGMA取0.1,或取更为严格的0.05;
※ 对于气体分子、原子体系:ISMEAR=0、SIGMA=0.01。
实际操作中的一个测试标准:SIGMA的取值要保证OUTCAR中的entropy T*S一项平均到每个原子上,要小于1-2meV。
1 | grep 'entropy T' OUTCAR |
IBRION 参数
IBRION参数决定了结构的优化过程。一般而言,优化结构的时候常用的参数有:
※ IBRION=3,当初始结构很差的时候;
※ IBRION=2,共轭梯度算法,一般最常用的选择;
※ IBRION=1,用于小范围内稳定结构的搜索。
ISPIN 参数
ISPIN的取值为1或2:
1:代表不考虑自旋极化(default);
2:代表打开自旋极化选项,在计算中考虑。
需要考虑自旋极化的几种常见情况:
※ 单原子计算
※ O2分子(基态为三重态)
※ 自由基相关计算
※ 含有Fe,Co,Ni的体系
※ 要计算的体系具有磁性
※ 关注体系的电子性质时
三重态氧分子:
首先回顾一下量子化学的相关知识,两个氧原子的原子轨道线性组合之后得到分子轨道,分子轨道又根据重叠程度的大小分为能量较低的成键轨道和反键轨道(*)。
对于氧分子而言,各个分子轨道的能量从低到高排列顺序为:
依据能量最低原则, 两个轨道上会占据有两个自旋方向相同的电子,使得氧分子的磁量子数s成为1,自旋多重度(2s+1)为3,故称为三重态氧分子。
MAGMOM 参数
MAGMOM参数用于指定体系中原子的初始磁矩,在计算复杂体系时有助于加快计算速度,并保持计算结果的正确性。
1 | MAGMOM = [real array] |
*注意:前后不应有空格
MOGMOM设置时,初始值不要求与实验值完全一致,一般来说取大一些(如1.5倍)比较好。
EDIFF 参数
控制电子步(自洽)的收敛标准,收敛速度与EDIFF的值呈指数关系。在大多数情况下,1E-4(默认值)足以胜任。
EDIFFG 参数
控制离子步(结构几何优化)的收敛标准:
※ 使用力作为收敛标准,此时EDIFFG通常设置为负值,取-0.01到-0.05之间;
※ 对于较大的体系,也可以使用能量作为标准,此时EDIFFG设置为正值,取0.0001-0.001。
NSW 参数
NSW控制几何结构优化的步数(即离子步),默认值为0,即为不进行任何结构优化。
※ 必须是大于等于0的整数;
※ 一般来说,简单的体系在200步以内即可正常结束;
※ 通常先设置较大值进行尝试。
NELM 参数
NSW控制电子自洽场计算的迭代步数(即电子步)。倘若计算中发现第一个离子步中的能量就不收敛,可以尝试增加NELM参数的值。
※ 对于一般的体系通常设置NELM=100;
※ 对于电子结构较难收敛的体系,可以设置NELM=200;
※ 倘若增加电子步效果不佳,可以尝试改变AMIX、BMIX两个参数。
1 | AMIX = 0.2 |
POTIM 参数
POTIM设置分子动力学的时间步长或离子弛豫的步宽(可以理解成离子步之间空间坐标的调整大小)。
※ 当IBRION=0时,VASP执行分子动力学模拟(MD)的任务,需设置模拟时间步长,必须提供;
※ 当IBRION=1,2,3时,POTIM默认设置为0.5;
※ 当IBRION=5,6时,POTIM默认设置为0.015。
当出现不收敛的情况是,可以根据当前IBRION设置下的默认值适当缩小POTIM来使计算更易收敛。
ENCUT 参数
ENCUT参数指定了展开波函数用到的平面波基组所对应的截断能量。
截断能越大,代表由于描述波函数的基组数量越多,计算精度越高,而相应的计算耗时也越长。
每个元素的赝势文件中都规定了元素价层电子与对应的最大截断能ENMAX。如果没有设置ENCUT,则会默认ENCUT=ENMAX。
ENMAX参数的获取方式:
1 | grep 'ENMAX' POTCAR |
在计算吸附等涉及多个体系的情形时需要注意ENCUT取值和KPOINTS设置的统一
NCORE/NPAR 参数
NCORE/NPAR参数用于控制并行计算:
※ NCORE:控制参与单个并行计算的核数量;
※ NPAR:控制并行计算的并行数。
NCORE=计算过程中划配的核数/NPAR
这两个参数只能选取其中之一来使用
ISIF参数
ISIF参数的可选设置值为1-7,其各自代表的意义如下表所示:
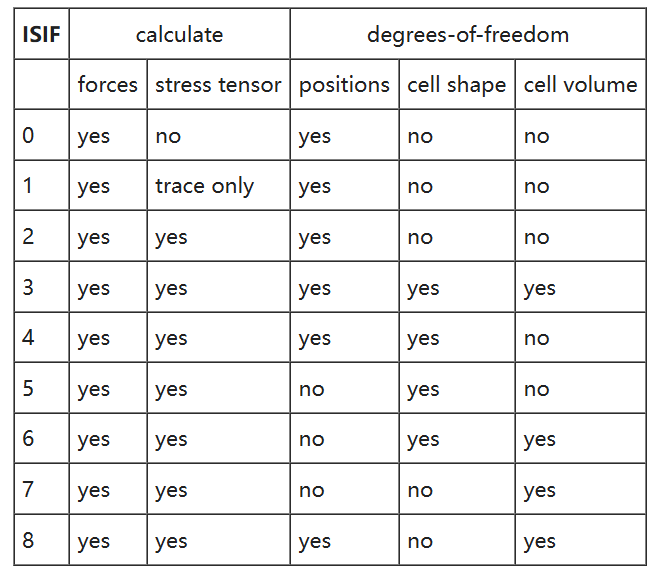
ISIF参数的默认值为2,选取不同的ISIF参数将在几何优化的过程中给体系设置不同的自由度。
例如ISIF=3时,对应了原子坐标、晶胞形状、晶胞体积均能够变化的情况。
当使用ISIF=3时,INCAR文件中必须设置ENCUT参数。
ALGO参数
电子最小化的算法,即电子波函数的优化算法。
※ normal:即矩阵对角化时先分割成子空间做对角化,能够得到稳定的不函数,不会发散;
※ VeryFast:RMM-DIIS算法;
※ Fast:先使用normal算法,再使用RMM-DIIS算法,前期保证收敛,后期加快速度;
※ All:波函数的所有带同时更新;
※ Damped:damped velocity friction 算法。
使用中normal和Fast两个算法最为常用。
LDIPOL参数
偶极子的存在与周期性边界条件的结合导致了总能量随超胞大小的缓慢收敛,进而导致势能和力的误差。上下不对称的slab表面所产生的偶极矩就属于这一情况。
这一问题可以通过设置LDIPOL=. true来抵消。
对于带电体系,需要设置LDIPOL时需要保证体系为立方体。
IDIPOL参数
控制计算的单极、偶极以及四极修正,取值为1-4:
※ IDIPOL = 1-3:仅计算单个方向上的修正,1-3对应x,y,z(第1-3晶矢)方向,常用于slab的计算;
※ IDIPOL = 4:所有方向上的修正都进行计算,常用于孤立分子的计算。
LWAVE参数
控制计算结束时是否将波函数写入WAVECAR文件:LWAVE = .FALSE(不写入)。
LCHARG参数
控制计算结束时是否将电荷密度写入CHGCAR文件:LCHAG = .FALSE(不写入)。